Abstract 1788: Mechanism of resistance to c-Met tyrosine kinase inhibitors in human melanoma
http://cancerres.aacrjournals.or ... etingAbstracts/1788
The over-expression of c-Met and activation by its ligand, hepatocyte growth factor (HGF), are known to cause significant tumor development and metastasis in human melanoma. Hence, c-Met is an attractive target for molecular therapeutic inhibition. To assess c-Met inhibition, we studied the effects of c-Met tyrosine kinase inhibitors (TKIs) in RU melanoma cells and found that JNJ38877605 and SU11274 (c-Met TKIs) inhibited cell growth in RU cells with an IC50 of 0.5 and 1µM, respectively. We then tested the therapeutic effects of JNJ38877605 in vivo. Five million RU melanoma cells were injected subcutaneously into the hind flanks of nude mice. Tumors were allowed to develop for a week after which daily oral treatments of 20 mg/kg JNJ38877605 or vehicle were given. We found that JNJ38877605 decreased the size of RU tumor xenografts in nude mice by 6 fold compared to mice treated with a vehicle control. Unfortunately, while c-Met inhibitors have been successful in clinical trials, acquired resistance to c-Met TKIs (ARQ197) and antibodies (MetMab) has been observed in cancer patients, resulting in tumor recurrence or lack of tumor CR. To determine the mechanism of c-Met TKI resistance, two melanoma cell line models, MU and RU, were exposed to increasing concentrations of SU11274. They proliferated in 7-10 fold higher concentrations of SU11274, and were cross resistant to 12 fold higher concentrations of JNJ38877605. After generating resistant cells, we measured HGF production and the expression levels of p-c-Met (Y1003) and p-c-Met (Y1234/1235) by immunoblotting to compare parental and resistant cells. We found that MU resistant cells exhibited upregulation of p-c-Met (Y1003) (6 fold) and p-c-met (Y1234/1235) (4 fold) compared to parental cells after treatment with HGF. Additionally, p-c-Met (Y1234/1235) was stable for only 30 min in parental cells compared to 60 min in resistant cells, where it was upregulated by 3-fold. This might be due to defects in c-Met ubiquitination resulting in its being recycled back or stabilized onto the membrane. Interestingly, we found that RU parental cells secreted 2 fold more HGF than the resistant cells. This indicates that resistant RU cells may not be as dependent on HGF induced activation in comparison to parental cells. In MU and RU resistant cells, we also found upregulation of p-mTOR (2 and 8 fold, respectively). Upregulation of phospho-pS6kinase (2 fold) and p-4E-BP1 (4 fold), downstream targets of p-mTOR, were also seen in MU cells. MU resistant cells also expressed increases in active β-catenin (protein downstream of Wnt signaling pathway) by 5 fold under no HGF stimulation and 2 fold after 60 min of HGF treatment. Our data indicates that the oral c-Met TKI, JNJ38877605, could be a promising therapeutic option for treating HGF producing melanoma tumors, but its effects could be further enhanced by the addition of mTOR and/or Wnt inhibitors to counteract acquired c-Met TKI resistance.
|
 第四代肺癌靶向药来袭,能否成为患者
作者:Tony
ALK 突变被称为肺癌的「钻石突变」,在非小细胞肺癌(NSCLC)中大约占5%,
第四代肺癌靶向药来袭,能否成为患者
作者:Tony
ALK 突变被称为肺癌的「钻石突变」,在非小细胞肺癌(NSCLC)中大约占5%,
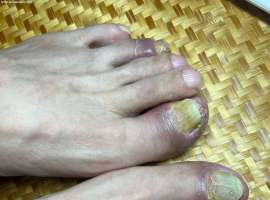 【求助】截至2024.8月一年半多使用k
68岁,男性,2022-11-21确诊为低分化肺腺癌IIIB期(T2N3M0)。截至2024年8月,共使用k
【求助】截至2024.8月一年半多使用k
68岁,男性,2022-11-21确诊为低分化肺腺癌IIIB期(T2N3M0)。截至2024年8月,共使用k
 母亲奥西耐药后应如何选择
2022年10月确诊肺腺癌,基因检测结果EGFR21,服用奥希替尼。2024年6月CT影像检查缓慢
母亲奥西耐药后应如何选择
2022年10月确诊肺腺癌,基因检测结果EGFR21,服用奥希替尼。2024年6月CT影像检查缓慢
 是进展了吗?下一步要怎么办
一线培美卡铂K四次,进展后换药
二线紫杉醇顺铂K六次,又有部分进展了还能怎么办啊
我
是进展了吗?下一步要怎么办
一线培美卡铂K四次,进展后换药
二线紫杉醇顺铂K六次,又有部分进展了还能怎么办啊
我
 我家的抗癌记录(四): 探索无人区—尝
迟迟无法动笔这一篇,实在因为在这段治疗期间所经历的艰辛,波折和痛苦,都不忍回想。
我家的抗癌记录(四): 探索无人区—尝
迟迟无法动笔这一篇,实在因为在这段治疗期间所经历的艰辛,波折和痛苦,都不忍回想。






 显身卡
显身卡