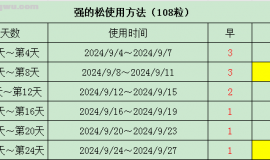
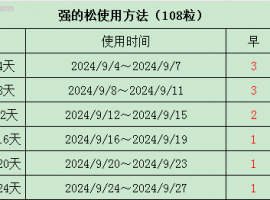
本帖最后由 十年后还在论坛 于 2015-6-11 19:38 编辑
NSCLC驱动基因:EGFR
活化EGFR突变位于酪氨酸激酶域,可导致组成性的EGFR信号。EGFR突变激活的PI3K-AKT 和 RAS-MEK-ERK信号对癌细胞的生长、生存和迁移起至关重要的作用。最常见的激活突变是19号外显子的框内缺失(in-frame deletion)同突变和858密码子的一个错义突变(导致精氨酸被亮氨酸取代,L858R)。带有EGFR突变的肺癌EGFR酪氨酸激酶抑制剂(TKIs)高度敏感。当前,EGFR突变的基因型筛查常被用于筛选患有IV期NSCLC、一线治疗方案为接受EGFR—TKIs治疗的患者。目前的研究重点集中在延长反应持续时间,找到有效的途径靶向在疾病进程中形成的耐药机制。最常见的耐药机制就是EGFR T790M突变,存在约50%的耐药肿瘤中。此外还有一些其他的例如MET扩增,PIK3CA突变以及向SCLC转化也曾得到描述。
NSCLC驱动基因:ALK
2号染色体的倒位导致生成了EML4-ALK融合基因,其编码的融合蛋白形成非配体依赖性二聚体引起组成性的ALK激活。ALK信号可通过激活RAS-MEK-ERK, JAK3-STAT3和PI3K-AKT 信号通路导致细胞增殖和生成。在NSCLC中ALK易位与腺癌组织学、印戒细胞形态学、年轻患者及非吸烟史相关。对于ALK抑制剂crizotinib的大型I期研究证实在包含ALK易位的癌症患者中整体反应率为57%,疾病控制率为90%,由此获得了FDA的批准。更有效的ALK抑制剂和靶向获得性耐药的策略目前还正在研发当中。
NSCLC驱动基因:ROS1
在1.5%的肺腺癌中存在有包含ROS1基因的染色体重排。与ALK阳性癌症相似,ROS1阳性癌症患者往往都比较年轻,无吸烟史,并患有腺癌。包含ROS1易位的癌症患者对于crizotinib的反应已经获得确定。
NSCLC驱动基因:KRAS
KRAS是肺癌中一种最常见的突变基因,发生在大约25%的肺腺癌中。肺癌KRAS突变主要定位在第12和13号密码子。肺癌中的KRAS突变似乎与EGFR和ALK易位互不相容,而患者通常都有吸烟史。KRAS突变通常抵抗EGFR-TKI治疗。虽然KRAS的发现比EGFR早二十多年,但由于研发难度大,目前针对KRAS的靶向药物少之又少,在临床实验阶段的仅有Antroquinonol(安卓健) 和AZD6244。其中AZD6244主要作用于RAS基因下游调控因子MEK1/2,而Antroquinonol(安卓健)直接作用于RAS基因,为该通路中讯息传递上游因子,调控整个RAS通路。其I期临床结果被证实有效且事直接作用于RAS的抑制剂,Antroquinonol(安卓健)有望在2015年作为非小细胞肺癌的常规治疗用药。
NSCLC驱动基因: PI3K
PIK3CA突变集中在两个热区,第9号和20号外显子,分别编码蛋白的螺旋域和激酶域。这些突变导致了脂质激酶活性增强,以及组成性的PI3K-AKT信号通路。目前有多个PI3K抑制剂正在研发中,其范围涵盖双PI3K/MTOR抑制,泛PI3K抑制以及亚型选择性PI3K抑制剂。临床前数据表明包含PIK3CA激活突变的癌症对单剂PI3K信号抑制剂最敏感,目前正在肺癌中开展临床试验验证这一推测。
NSCLC驱动基因: PTEN
肿瘤抑制基因PTEN编码的脂质磷酸酯酶对PI3K-AKT信号起负调控作用,PTEN丧失可导致组成性PI3K-AKT信号。PTEN在大量癌症中可通过多种机制失活。相比于腺癌,PTEN丧失在鳞状细胞癌中更常见。临床试验正在评估PI3K抑制剂在PTEN缺失癌症中的效应。
NSCLC驱动基因:FGFR1
FGFR1是一个潜在的肺鳞状上皮细胞癌靶标。FGFR1是RTKs家族FGFR成员。FGFR1激活可导致通过PI3K-AKT 和RAS-MEK-MAPK的下游信号。在大约20%的鳞状细胞癌中发现有FGFR1扩增。在实验室研究中,抑制包含FGFR1的癌细胞系和小鼠模型中的FGFR1可导致生长抑制和凋亡。多种FGFR抑制剂正在临床研发阶段,其中许多抑制剂除了FGFR1还可抑制多种酪氨酸激酶。
NSCLC驱动基因:PDGFRA
在肺鳞状细胞癌中发现有PDGFRA的扩增。通过shRNA敲除或小分子抑制PDGFRA可损害细胞存活和贴壁非依赖性的生长,表明PDGFRA可能是带有PDGFRA扩增的癌症的一种驱动癌基因。多种PDGFR抑制剂目前在临床开发中。与FGFR1抑制剂相似,大量的药物都能抑制多种激酶。
NSCLC驱动基因:DDR2
DDR2是一种可与胶原蛋白结合的受体酪氨酸激酶(PTK),可促进细胞迁移、增殖和存活。DDR2突变存在于肺鳞状细胞癌和细胞系中。在DDR2突变细胞系中,抑制DDR2活性可导致增殖抑制。异位表达突变DDR2可导致细胞转化,而不同的突变导致的转化水平有所差异。这些结果表明DDR2突变可能是致癌性的,带有这些突变的癌症可能会对DDR2激酶抑制剂敏感。
NSCLC驱动基因:BRAF
BRAF突变存在于1%–3%的非小细胞肺癌中。V600E是最常见的突变,在肺癌中也有多种其他类型的BRAF突变被报道,包括G469A和D594G。尽管特异性药物例如vemurafenib在包含BRAF V600E突变的黑色素瘤中高度有效,但这些药物在BRAF突变肺癌中的活性还需评估。多项评估BRAF和MEK抑制剂活性的试验正在BRAF突变的癌症中开展。
肺腺癌的驱动基因
EGFR突变是NSCLC最常见的驱动基因,约10%的高加索NSCLC患者和30-40%的东亚NSCLC的患者存在EGFR突变,EGFR突变在不吸烟腺癌患者中发生率较高。大量临床研究已经证实以EGFR-TKI一线治疗EGFR突变的晚期NSCLC患者的疗效优于化疗,NCCN指南推荐EGFR突变患者一线使用EGFR-TKI。另一驱动基因ALK重排从2007年第一次发现到它的第一个酪氨酸激酶抑制剂Crizotinib,批准进入临床仅仅用了不到5年时间,目前,Crizotinib,已被NCCN指南推荐作为ALK重排患者的一线治疗。靶向药物在肺腺癌中的迅猛发展令人鼓舞,许多除EGFR和ALK以外的驱动基因也不断地被发现并有可能成为下一个有效治疗靶点:
1. KRAS和NRAS
KRAS突变存在于15%一20%的NSCLC。KRAS是RAS GTP酶家族的成员之一,可以通过Ras/Raf信号通路促进细胞的生长分化。这些酶通过与GTP结合,发挥RAS家族的GTP酶活性,使GTP转化为GDP,从而使下游信号瀑布中的蛋白发生磷酸化。当KRAS发生突变时(主要发生于外显子12[80%],13和61)降低了KRAS作为GTP酶的活性,使其具有致瘤的特性。到目前为止KRAS被认为是预示化疗和靶向治疗疗效不佳的预测因子而非一个有效的药用靶点:与结直肠癌不同的是,在NSCLC中KRAS突变与抗EGFR通单克隆抗体耐药的相关性并不明确:突变的KRAS与GTP的高亲和性限制了直接抑制KRAS的药物的研发和应用。直到20l3年,一种能与KRAS突变异构体结合的KRAS G12C抑制剂被报道,但这种药物的临床应用还有很长的路要走。目前对于KRAS突变NSCLC患者的治疗策略主要集中于干扰其下游信号通路,如PI3K,MEK和FAK,都还处于临床研究阶段。最具有临床应用前景的治疗策略是细胞毒性化疗药物与MEK抑制剂的联合应用:多西他赛与口服MEK抑制剂Selumetinib联合治疗KRAS突变的NSCLC被证实在临床前模型中有效,并且在一项Ⅱ期临床研究中显示多西他赛联合Selumetinib疗效优于多西他赛单药,患者的有效率提高,PFS长。这种联合治疗目前正在处于注册临床研究的评估阶段(SELECT-1, NCT01933932)。其他RAS家族成员包括HRAS和NRAS。约l%的NSCLC患者存在NRAS体细胞突变。NRAS突变在腺癌及有吸烟史的肺癌患者中发生率较高,并在临床前模型中NRAS突变的NSCLC显示出对MEK抑制剂敏感。
2. ROS1
ROS1是胰岛素受体家族的一种受体酪氨酸激酶。ROS1 重排最早在胶质母细胞瘤中被发现,位于6号染色体上。近年来, ROS1融合基因被认为是NSCLC的驱动基因,约在1%-2%的NSCLC中检测到ROS1重排。ROS1包含了完整的酪氨酸激酶域,49%的激酶域和77%的ATP结合位点与ALK存在高度的氨基酸同源性,发生基因融合后可以导致下游细胞生长和增殖相关信号通路活化,ROS1融合基因阳性的NSCLC患者通常有年轻的不吸烟腺癌的临床特征,ALK/MET/ROS1多靶点抑制剂Crizotinib被证实对ROS1融合基因阳性的患者有效。在PROFILE1001显示,每日两次口服250mg Crizotinib治疗ROS1融合基因阳性患者,有效率和疾病空置率分别达到57%和79%。
3. BRAF
BRAF属于MAPK信号通路的丝氨酸苏氨酸蛋白激酶家族。约1%-3%的NSCLC存在BRAF突变。这些携带BRAF 突变的NSCLC患者通常为吸烟/曾吸烟的人群,并且与其他突变相互排斥,通常不同时存在。与黑色素瘤不同的是在肺癌中,研究者检测到了BRAF的多种突变位点,如V600E(50%)、G469A(40%)、D594G(11%),而不是像黑色素瘤一样以V600E为主。许多BRAF抑制剂,包括 sorafenib、vemurafenib 和dabrafenib正在临床研究阶段。一项BRAF V600E特异性抑制剂vemurafenib治疗BRAF V600E突变的晚期实体肿瘤的II期临床实验已经开始。对于非V600E类型BRAF突变, 使用V600E特异性抑制剂治疗是无效的,但针对下游的靶向药物如MEK抑制剂是否有效正处于探索阶段。
4. HER2(ERBB2)
人表皮生长因子受体2(HER2)是ERBB家族的一员,虽然HER2没有已知的配体与之结合,但其可以与ERBB家族的其他任一成员结合成为二聚体。约2%-4%的NSCLC患者存在HER2突变,多数患者具有不吸烟腺癌的临床特征,最常见的突变类型是发生于20外显子的插人突变A775_G776 ins YVMA,而在EGFR/KRAS/ALK均阴性的NSCLC穿刺标本中,HER2突变的发生率为6%。20外显子插人突变导致HER2激酶活性增加,从而激活下游信号通路,促进细胞增殖转移及致瘤性。相对于乳腺癌和胃癌,HER2扩增并没有在肺癌中显示出具有提示较好预后的作用。HER2突变患者用HER抑制剂可能获得较好的疾病控制率,在细胞实验中也证实HER2 20外显子突变的细胞对EGFR/HER2双靶点药物,如neratinib、dacomitinib和afatinib 敏感。
在一项I期临床试验中,6位携带HER2 20外显子插人突变的NSCLC患者使用HER2抑制剂neratinib联合mTOR抑制剂temsirolimus进行治疗,其中2位患者部分缓解。另一项小规模的Ⅱ期临床试验中,3位HER2 20外显子插入突变的晚期NSCLC患者经afatinib治疗均获得部分缓解。在一项回顾分析中,4位服用Afatinib单药治疗的HER2 20外显子插入突变的NSCLC患者的疾病控制率达100%,然而另外2位服用Lapatinib单药的HER2插入突变患者均疾病进展。在这项回顾性研究中,还发现15位HER2 20外显子突变的NSCLC患者用trastuzumab联合化疗的疾病控制率达 96%。而这些HER2抑制剂治疗HER2突变NSCLC的诸多临床试验也正在如火如荼地进行中。
5. RET
RET基因编码RET受体酿氨酸激酶,约1%的NSCLC患者存在RET基因重排。CCDC6-RET, KIF5B-RET和TRIM33-是已被发现的三种RET融合基因型。RET重排目前只在腺癌中检测到,并且不与EGFR突变ALK重排,KRAS突变等同时存在。许多已上市的多靶点酪氨酸激酶抑制剂有抑制RET的活性,在体外实验证实重排的肿瘤细胞对vandetanib、Sorafenib、Sunitinib、Cabozantinib敏感,但这些药物对有RET重排肺癌患者的疗效还不能完全肯定。虽然到目前为止,针对RET融合基因阳性患者对何种治疗效果较好的回顾性和前瞻性研究还非常有限,但有一项用Cabozantinib治疗RET重排NSCLC患者的单臂Ⅱ期临床实验正在进行,其结果非常值得期待。
6. NTRK1
NTRK1是编码高亲和性神经生长因子(TRKA蛋白)的基因,就目前的研究发现约3%的肺腺癌存在NTRKD的重排,且不与EGFR突变KRAS突变和ROS1融合基因同时存在。目前发现的NTRK融合基因型为MPRIP-NTRK1和CD74- NTRK1,这两种基因型在体外实验中被证实可以TRKA蛋白发生自磷酸化从而激活其致瘤作用。在体外模型中,具有 抗TKRA活性的酪氨酸激酶抑制剂ARRY-470, lestaurtinib(CEP-701)和Crizotinib可以使细胞周期停滞,抑制细胞增殖。在Vaishnavi的研究中,携带MPRIP-NTRK1的NSCLC患者接受了Crizotinib的治疗,该患者在接受治疗后肿瘤缩小,CA125下降,但在3个月后疾病进展。虽然目前还没有专门针对肺癌NTRK1融合基因阳性患者的临床试验,但TSR-011、 PLX7486等具有TRK抑制剂作用的药物在NRTK重排的实体瘤中的临床试验已经开始。
7. MEK1
MEK1是BRAF下游增殖信号通路的丝氨酸苏氨酸激酶,约1%的NSCLC存在MEK1突变,这种突变在肺腺癌中较肺鳞癌多见,主要突变位点K57N,Q56P和D67N。在体外模型中,MEK1突变可以导致信号通路持续激活并对MEK抑制剂敏感。但MEK抑制剂在临床中应用的疗效目前还未知晓,一项MEK抑制剂MEK162的II期临床试验已在RAF, RAS, NF1或MEK突变的实体瘤患者中展开。
8. MET
MET是一种受体酪氨酸激酶,与其配体肝细胞生长因子结合而激活。约25%的NSCLC存在MET蛋白的过表达,并与不良预后相关。扩增约存在于2%-4%的肺腺癌和肺鳞癌,MET扩增也与NSCLC的不良预后相关。在EGFR突变的EGFR-TKI获得性耐药NSCLC患者中,约20%的患者存在扩增。MET扩增的NSCLC患者可能对MET抑制剂敏感,许多MET的酪氨酸激酶抑制剂(如Crizotinib, Tivantinib等),以及MET的单克隆抗体(如Onartuzmnab等)的临床试验正在MET扩增或MET蛋白过表达的肿瘤患者中展开。
二、肺鳞癌的驱动基因
尽管研究者在寻找肺鳞癌驱动基因上做了很多努力,但肺鳞癌分子标志物的研究步伐远落后于腺癌,肺腺癌的驱动基因很少在肺鳞癌中被检测到。并且一些较新的药物例如贝伐单抗和培美曲塞也不支持用于肺鱗癌或被证实疗效不理想。因此,存在晚期肺鱗癌患者较非鱗癌患者的治疗选择要少得多。然而,最近越来越多的研究发现驱动基因突变也存在于肺鱗癌,并且可能与肺鱗癌的靶向治疗疗效相关,这些基因包括FGFR1,DDR2和PIK3CA等。
1. FGFR1
成纤维细胞生长因子受体(fibroblast growth factor receptor, FGFR)包括四种受体酪氨酸激酶(FGFR1、FGFR2、FGFR3和FGFR4)。FGFR1扩增约占肺鱗癌的20%,但在肺腺癌中仅约2%的患者检测到FGFR1扩增。另外,5%~10%的肺鱗癌患者存在FGFR2/3/4扩增或突变,这些基因的改变通过激活MAPK和PI3K信号通路促进细胞增殖。有关FGFR抑制剂 (例如AZD4547、JNJ-42756493、BGJ398、DoVitinib、Ponatinib 等)的I~Ⅱ期临床研究已经在肺鱗癌中展开。
2. DDR2
盘状死亡受体(Discoidin death receptor 2,DDR2)是一种只能被胶原激活而非肽类生长因子激活的酪氨酸激酶受体,它可以激活SRC和STAT信号通路促进细胞增殖。DDR2突变约在4%的肺鱗癌中被检测到,而在非鱗肺癌中发生的比例不到1%。多靶点酪氨酸激酶抑制剂达沙替尼(Dasatinib)可以抑制DDR2,并已在裸鼠荷瘤模型中证实达沙替尼能够抑制DDR2突变的NSCLC,同时在一位使用厄洛替尼和达沙替尼联合治疗使肿瘤明显缩小的肺鳞癌患者中,研究者检测到该患者肿瘤组织存在DDR2激酶域S768R突变。在一则病例报道中,一位患有慢性髓细胞性白血病和DDR2 S768R突变的肺鱗癌患者对达沙替尼治疗有效。一项使用达沙替尼治疗DDR2突变晚期肿瘤的II期临床研究正在进行。
3. PI3KCA
鱗脂酰肌醇3激酶催化OL多肽(phosphoinositide-3-kinase,catalytic, a polypeptide, PIK3CA)基因编码PI3K的催化单元,是一种通过AKT/mTOR信号通路调节细胞生长与增殖的脂类激酶。PIK3CA突变通常发生在外显9和外显子20约1%~3%的NSCLC存在PIK3CA突变。在肺鱗癌和肺腺癌中的突变率相似,多数具有PIK3CA突变的患者有吸烟史。与其他类型的突变相比,P/K5C4突变与EGFR突变同时存在的概率相对较高,约5%的EGFR突变患者对EGFR-TKI发生获得性耐药时出现了PIK3CA突变。PI3K,AKT和mTOR抑制剂在PIK3CA突变NSCLC中的临床效果如何还未知晓,仍在临床试验进行过程。另有一项PI3K抑制剂Buparlisib与多西他赛联合治疗晚期肺鱗癌的Ⅱ期临床研究正在进行。 |
 神奇的ommaya囊——学习笔记
一个神奇的装置-ommaya囊——————9.28 苗茜医生科普讲座
直播链接:https://live.
神奇的ommaya囊——学习笔记
一个神奇的装置-ommaya囊——————9.28 苗茜医生科普讲座
直播链接:https://live.
 免疫性肺炎过程
免疫性肺炎过程:22次替雷利珠单抗用后,距最近使用时间(7月23日)15天,8月7日出现
免疫性肺炎过程
免疫性肺炎过程:22次替雷利珠单抗用后,距最近使用时间(7月23日)15天,8月7日出现
 肥胖促癌原因找到了!这种“酸”成为
作者:雨过天晴
常有人开玩笑说“一胖毁所有”,其实这是真的。肥胖已经成为一项影响
肥胖促癌原因找到了!这种“酸”成为
作者:雨过天晴
常有人开玩笑说“一胖毁所有”,其实这是真的。肥胖已经成为一项影响
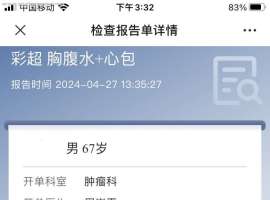 父亲的治疗传来了好消息
父亲今年68岁,从今年三月低确认肺癌晚期到现在,一直没有好消息。胸水基因检测:EGFR
父亲的治疗传来了好消息
父亲今年68岁,从今年三月低确认肺癌晚期到现在,一直没有好消息。胸水基因检测:EGFR
 求助 K药和替雷利珠如何选择
父亲9月刚查出非小细胞低分化癌伴随左肾上腺和第10胸椎转移PDL1高表达(TPS=90%) SMARC
求助 K药和替雷利珠如何选择
父亲9月刚查出非小细胞低分化癌伴随左肾上腺和第10胸椎转移PDL1高表达(TPS=90%) SMARC




 显身卡
显身卡